First-principles program using the all-electron mixed basis approach
TOMBO – Gallery
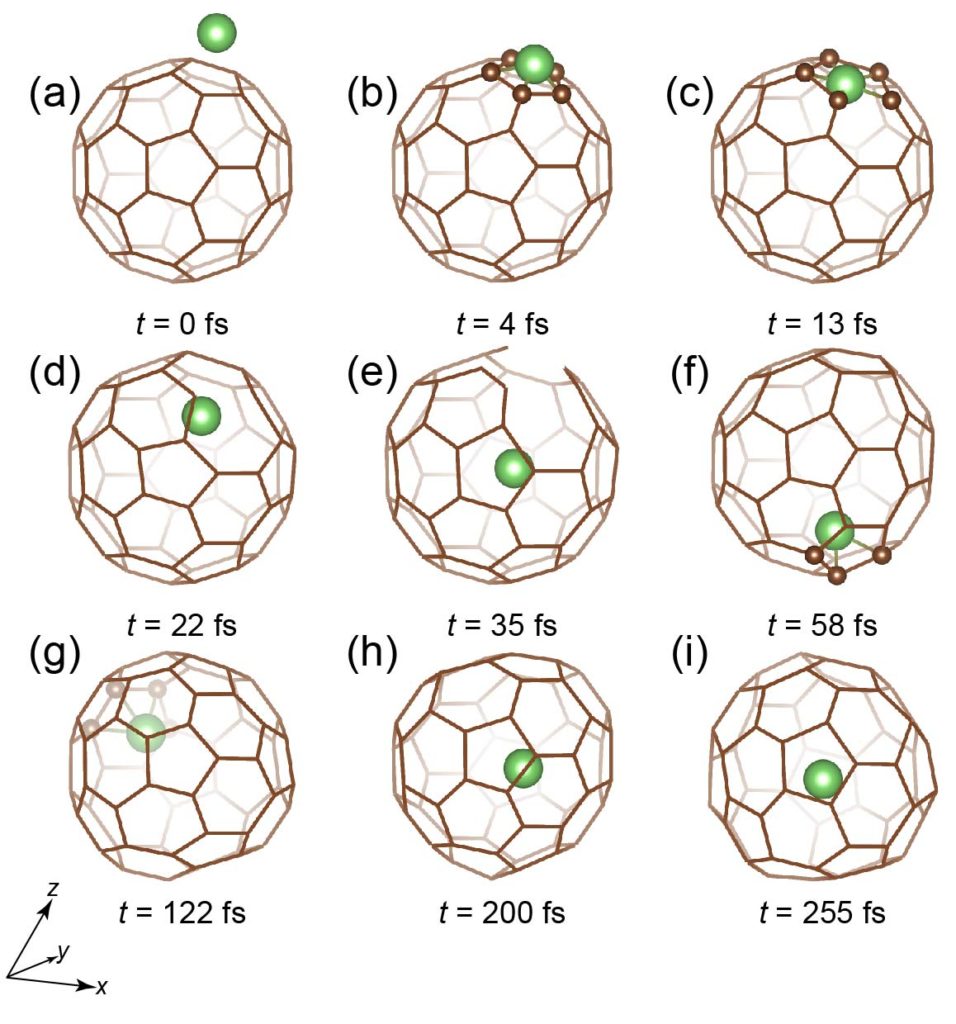
Li atom going through a five-membered ring of C60.
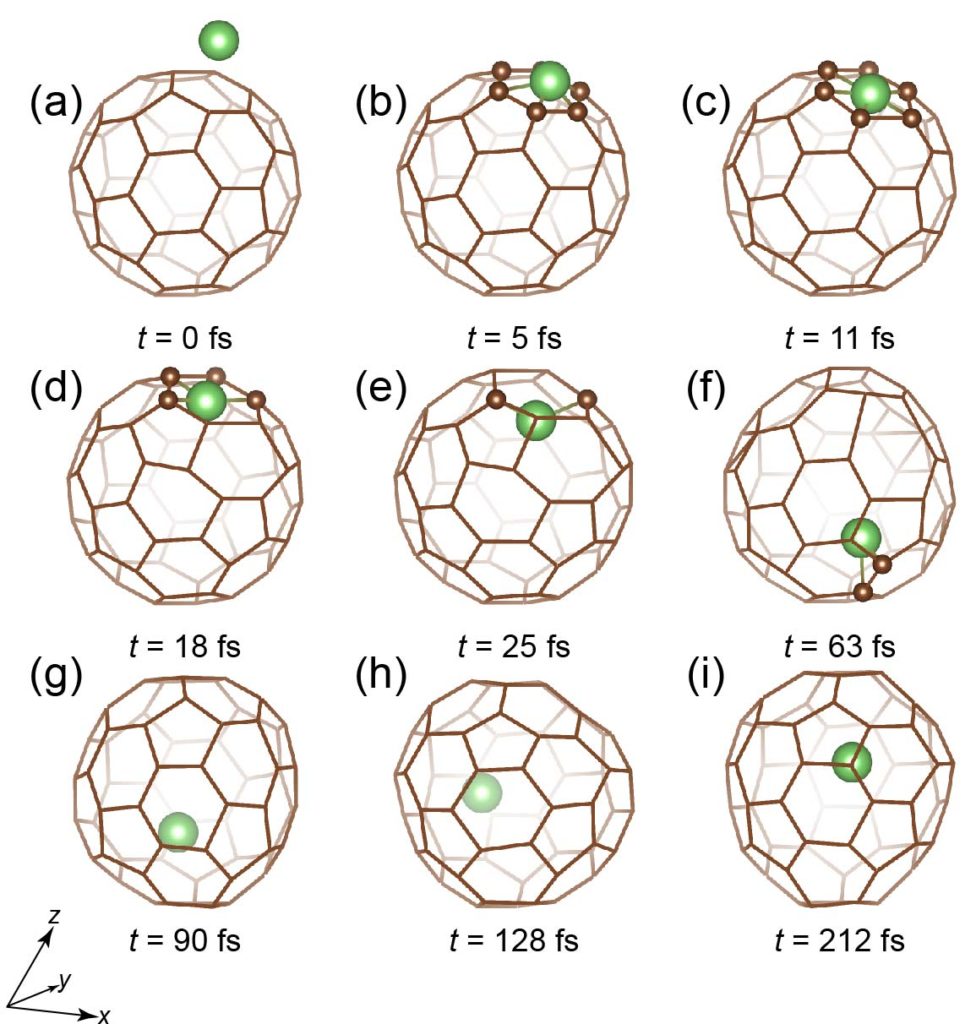
Li atom going through a six-membered ring of C60.
A first-principles molecular dynamics simulation of Li+ insertion through 0.5 Å off-center position of a six-membered ring of C60 with 30 eV kinetic energy in 10.5° inclined direction from the surface normal to be compared with the plasma shower experiment. The expected production ratio of the endofullerene Li+@C60 is 3.7 ± 0.5 % for 30 eV kinetic energy. (video produced by AVS)
K. Ohno, A. Manjanath, Y. Kawazoe, R. Hatakeyama, F. Misaizu, E. Kwon, H. Fukumura, H. Ogasawara, Y. Yamada, C. Zhang, N. Sumi, T. Kamigaki, K. Kawachi, K. Yokoo, S. Ono, and Y. Kasama, Nanoscale 10, 1825-1836 (2018). https://doi.org/10.1039/C7NR07237F
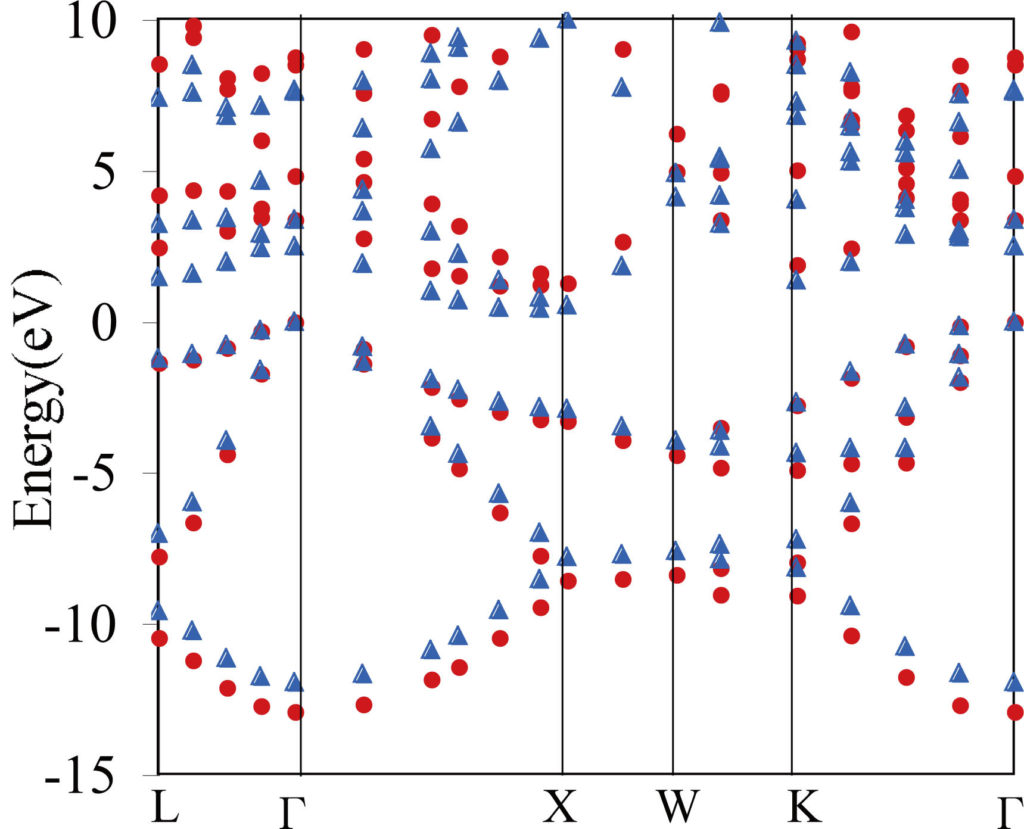
Si band calculated by LDA (▲) and GW (⚫️).
Ti0.75Nb0.25O2 band calculated by LDA (−) and GW (⚫️).
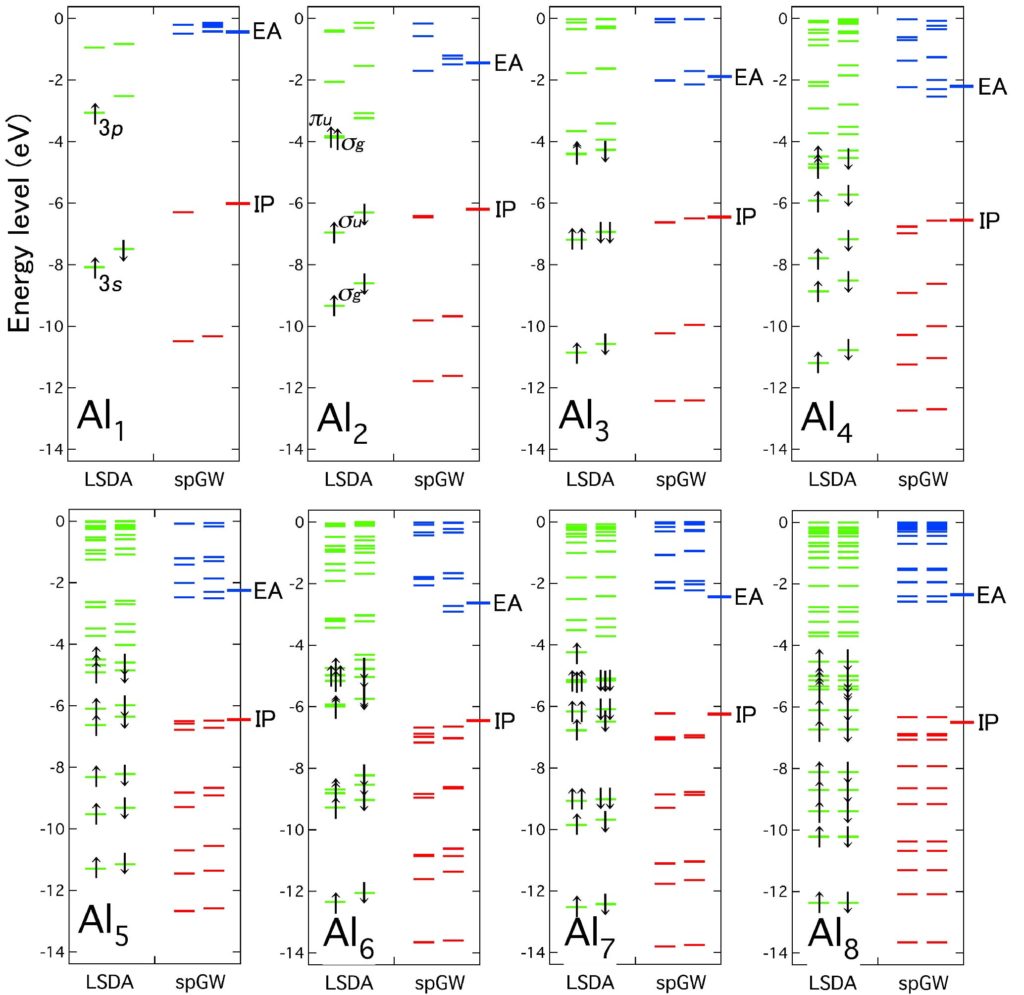
Quasiparticle levels calculated by LSDA and spin-polarized GW (spGW). On the right side of each pannel,− EA and − IP indicate minus of experimental electron affinity (EA) and ionization potential (IP), which are in excellent agreement with the lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) levels calculated by spGW.
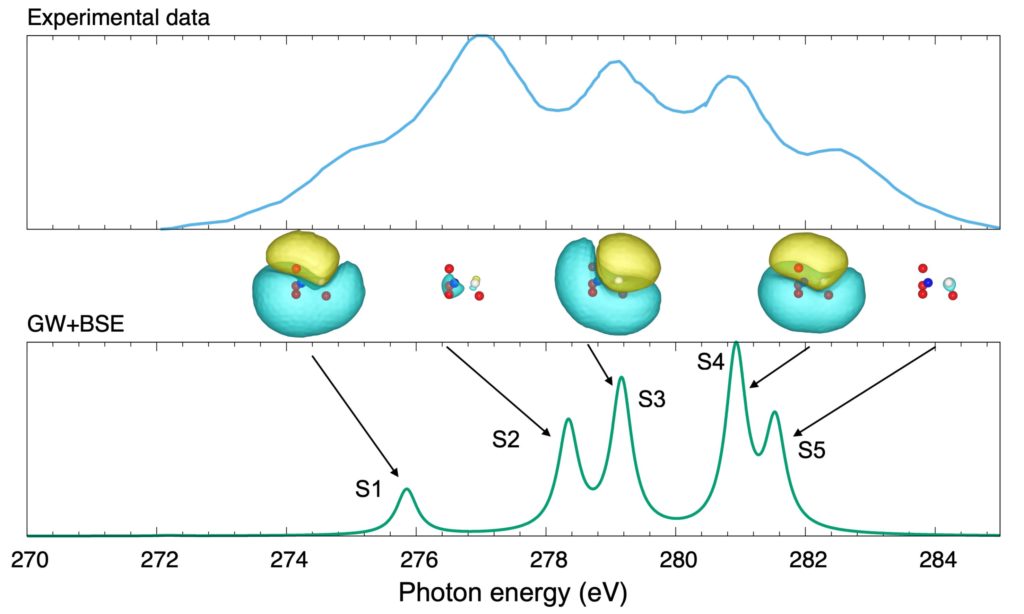
Experimental and calculated (GW + BSE) X-ray emission spectra (XES) of a methanol (CH3OH) molecule. The initial state is a highly excited eigenstate with a deep core hole created by preceding XPS spectroscopy (XPS). It is emphasized that this type of calculation is possible on the basis of extended quasiparticle theory (EQPT); see also Top page.